

-
Probing chiral recognition in liquid chromatography by absolute configuration modulation ATR-IR spectroscopy
R. Wirz, D. Ferri, T. Bürgi and A. Baiker
Spectroscopy Europe, 19 (1) (2007), p8-16


unige:14678
|
|
 |
|||||||
O-Phenylcinchonidine (PhOCD) is known to efficiently induce inversion of enantioselectivity with respect to cinchonidine (CD) in the enantioselective hydrogenation of various activated ketones on Pt/Al2O3. To understand the origin of the switch of enantioselective properties of the catalyst, the adsorption of PhOCD has been studied by in situ ATR-IR spectroscopy, in the presence of organic solvent and dissolved hydrogen, i.e., under conditions used for catalytic hydrogenation. The adsorption structures and energies of the anchoring group of CD and PhOCD were calculated on a Pt 38 cluster, using relativistically corrected density functional theory (DFT). Both approaches indicate that both modifiers are adsorbed via the quinoline ring and that the spatial arrangement of the quinuclidine skeleton is critical for the chiral recognition. New molecular level information on the conformation of CD relative to PhOCD adsorbed on a surface is extracted from the ATR spectra and supported by DFT calculations. The result is a clearer picture of the role played by the phenyl group in defining the chiral space created by the modifiers on Pt. Moreover, when CD was added to a pre-equilibrated adsorbed layer of PhOCD, a chiral adsorbed layer was formed with CD as the dominant modifier, indicating that CD adsorbs more strongly than PhOCD. Conversely, when PhOCD was added to preadsorbed CD, no significant substitution occurred. The process leading to nonlinear effects in heterogeneous asymmetric catalysis has been characterized by in situ spectroscopy, and new insight into a heterogeneous catalytic RâS switch system is provided. | ||||||||
|
||||||||
The behavior of ethyl pyruvate during adsorption on vapor deposited alumina-supported platinum films and on a commercial 5 wt % Pt/Al2O3 catalyst has been studied in the absence and presence of coadsorbed cinchonidine, which is usually applied as a chiral modifier in the platinum-catalyzed enantioselective hydrogenation of α-ketoesters. The in situ ATRâIR measurements, performed at room temperature using hydrogen-saturated CH2Cl2 as solvent, revealed that upon adsorption on the platinum some of the ethyl pyruvate decomposes leading to strongly adsorbed CO and other fragmentation products. The CO originating from decomposition of ethyl pyruvate reached approximately 14% of the amount of adsorbable CO on the free platinum surface and is proposed to be adsorbed preferentially on energetically favored sites such as edges and corners. The presence of cinchonidine (10-4 M) lead to a drastic decrease of the decomposition rate of ethyl pyruvate by a factor of about 60 under the conditions used. Competitive adsorption experiments of CO and cinchonidine in the presence of hydrogen indicated that cinchonidine can displace the adsorbed CO, confirming the strong anchoring of cinchonidine on the platinum surface, which is a prerequisite for its action as a chiral modifier. The findings of the adsorption studies provide a plausible explanation for the earlier made observation that the sequence of admission of α-ketoester, chiral modifier, and hydrogen affects the catalytic performance of platinum-catalyzed enantioselective hydrogenation. The decomposition is likely to occur also with other α-ketoesters and may have a bearing on the initial transient period, typically observed during hydrogenation of such compounds on cinchona-modified platinum catalysts. | ||||||||
|
||||||||
The interaction of 2-methoxypropene, a vinyl ether, with heterogeneous acid catalysts containing sulfonic acid groups covalently bound to SiO2 (Deloxan ASP, Degussa) and sulfuric acid adsorbed on TiO2-modified amorphous SiO2 (Degussa), respectively, was investigated by in situ attenuated total reflection infrared spectroscopy. Rapid hydrolysis is observed, which does not, however, require the acid sites. The resulting acetone is adsorbed predominantly on SiOH groups. Promoted by the acid sites a further transformation is observed on the catalysts. Based on the time behavior of the ATR signals of acetone and the product the further reaction likely involves the condensation of 2-methoxypropene and acetone. During the buildup of the reaction product hydronium ions disappear from the catalyst surface. Upon desorption of the reaction product the hydronium ions become prominent again on the catalyst containing adsorbed sulfuric acid. This behavior is less pronounced on the catalyst, which contains sulfonic acid groups. The two investigated catalysts contain vastly different relative concentrations of Brønsted and Lewis acid sites, which can explain the difference in the relative concentration of intermediate and product at the interface in the observed consecutive reaction. | ||||||||
 |
|
|||||||
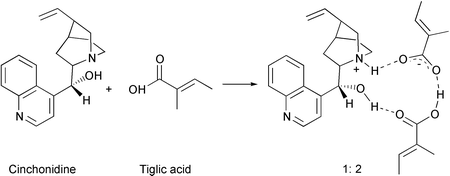
Cyclic cinchonidine ⶠacid complexes (1 ⶠ1 and 1 ⶠ2) of the chiral modifier cinchonidine (CD) and an alkenoic acid, tiglic acid, in dichloromethane solvent have been observed by FTIR spectroscopy. Both the OH and the quinuclidine N atom of CD are involved in the hydrogen bond with the acid molecule(s). Such dual-site modifierâreactant interactions play an important role in the enantioselective hydrogenation of alkenoic acids over CD-modified Pd catalysts. The stability of these 1 ⶠ1 and 1 ⶠ2 complexes has been probed by addition of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), a stronger base than CD. DBU builds ion pairs with the acid (with 1 ⶠ1 and 1 ⶠ2 stoichiometry) and a hydrogen bond with the OH of CD. However, despite the large difference in basicity between CD and DBU, 1 ⶠ2 CD ⶠacid complexes can still be detected when more than 0.5 equivalent DBU was added with respect to the acid, at which ratio the enantiomeric excess (ee) drops dramatically. Hence, the molecular structure of CD favours formation of cyclic complexes via a dual-site interaction, which is not possible for DBU ⶠacid complexes, and stabilises 1 ⶠ2 CD ⶠacid species, which are proposed to be responsible for enantiodifferentiation. | ||||||||
|
||||||||
|
||||||||
Pd/Al2O3 model catalysts have been prepared by physical vapour deposition and characterised by means of XPS, STM, and in situ ATRâIR spectroscopy. Morphological changes in the Pd film induced by dissolved hydrogen leads to enhanced infrared absorption and could be followed with both STM measurements and IR spectroscopy. Adsorption of CO, pyridine, quinoline, 2-methylquinoline, and the chiral auxiliary cinchonidine has been studied in situ at 283 K in CH2Cl2 solvent. Two different species have been observed for cinchonidine on Pd. One is oriented with the quinoline moiety nearly parallel to the Pd surface, likely through the Ï-system, whereas in the second the Ï-bonding through the N lone pair prevails and induces a tilting of the ring with respect to Pd. No indication of the presence of α-quinolyl species has been found, in contrast to adsorption on Pt/Al2O3 catalysts. Compared to adsorption on Pt, cinchonidine is more weakly bound on Pd under hydrogenation conditions. Also, the relative stability of the Ï- and N lone pair-bonded species is different for the two metals, with the Ï-bonded species being relatively more stable on Pt. Similarities and differences found in the adsorption of the chiral modifier on the two metals are discussed and traced mainly to the different d-orbital diffuseness of Pd and Pt. | ||||||||
|
||||||||
The adsorption of carboxylic acids (formic, acetic, and pyruvic acid) from corresponding solutions in CH2Cl2 solvent on Al2O3 and TiO2 thin films has been studied by attenuated total reflection infrared spectroscopy. The metal-oxide films were vapor-deposited on a Ge internal reflection element, which was mounted into a specially designed flow cell. The system allowed in situ monitoring of the processes occurring at the solid-liquid interface. The metal-oxide films were characterized by X-ray photoelectron spectroscopy, ellipsometry, and atomic force microscopy. Formic acid and acetic acid adsorbed predominantly as bridging species on alumina surfaces. Adsorbed free acids were not observed under a flow of neat solvent. Based on the position of the νAS(COO) and of the keto-group stretching vibration of the pyruvate ion, pyruvic acid is proposed to coordinate to the Al2O3 surface in a monodentate fashion, whereas, on TiO2, a bidentate species is preferred. Comparison of the adsorption behavior on the vapor-deposited alumina film and on an α-Al2O3 layer deposited from a water suspension of the corresponding metal-oxide powder indicated that pyruvic acid adsorbs in a similar mode, irrespective of the metal-oxide deposition technique. | ||||||||
|
||||||||
Model platinum catalysts have been designed to study the platinumâsolvent interface in situ using attenuated total reflection (ATR) infrared spectroscopy. Pt and Pt/Al2O3 thin films were evaporated on a Ge internal reflection element (IRE) and characterized by XRD, XPS, AFM, STM, and IR spectroscopy. Changes within the adsorbate layer of the Pt catalyst during cleaning with O2 and H2 were followed. After cleaning, the catalyst surface was probed by CO adsorption from CH2Cl2. For the Pt/Al2O3 film the spectrum of adsorbed CO showed a band at 2000 cm-1, which is typical for Pt/Al2O3 catalysts. The stretching vibration of linearly bonded CO exhibited a coverage-dependent frequency shift due to vibrational coupling, thus showing the existence of large clean domains on the reactive catalyst surface even in the presence of an organic solvent. CO adsorption from CH2Cl2 was slow before the cleaning process. However, subsequent admission of H2 resulted in an instantaneous and drastic increase of the CO absorption signal. The origin of this effect is a structural change of the Pt particles induced by dissolved hydrogen, which was directly monitored by ATR spectroscopy using CO as probe molecule. STM investigations showed sintering of the Pt particles upon hydrogen treatment in CH2Cl2 at room temperature, which leads to a surface-enhanced infrared absorption (SEIRA). | ||||||||
|
||||||||
Adsorption of cinchonidine on a platinum model catalyst studied by in situ ATR-IR spectroscopy revealed that the adsorption mode depends on surface coverage and is affected by concomitant adsorption and fragmentation of solvent molecules. | ||||||||
|
||||||||
An in situ attenuated total reflection study of the chiral solidâliquid interface created by cinchonidine adsorption on a Pt/Al2O3 model catalyst is presented. Experiments were performed in the presence of dissolved hydrogen, that is under conditions used for the heterogeneous enantioselective hydrogenation of α-functionalized ketones. Cinchonidine adsorbs via the quinoline moiety. The adsorption mode is coverage dependent and several species coexist on the surface. At low concentration (10-6M) a predominantly flat adsorption mode prevails. At increasing coverage two different tilted species, α-H abstracted and N lone pair bonded cinchonidine, are observed. The latter is only weakly bound and in a fast dynamic equilibrium with dissolved cinchonidine. At high concentration (10-4â10-3 M) all three species coexist on the Pt surface. A slow transition from an adsorbate layer with a high fraction of α-H abstracted cinchonidine to one with a high fraction of N lone pair bonded cinchonidine is observed with the cinchonidine concentration being the driving force for the process. The reverse transition in the absence of dissolved cinchonidine is fast. Cinchonidine competes with solvent decomposition products for adsorption sites on the Pt, which may contribute to the observed solvent dependence of the heterogeneous enantioselective hydrogenation of ketones by cinchonidine-modified Pt. | ||||||||
|
||||||||
The conformational behaviour of several α-ketoesters was investigated using solution FTIR in combination with ab initio calculations. The α-ketoesters show marked differences in the O=CâC=O torsional potential energy surface depending on the substituent at the α-keto group. In general the torsional potential is characterised by broad minima corresponding to s-cis and s-trans conformations and low interconversion barriers. The s-trans conformation is more stable but the fraction of s-cis is considerable at room temperature and increases with solvent polarity due to the higher dipole moment of the latter. Hydrogen bonding with alcoholic solvents also leads to a stabilisation of the s-cis conformer. The interaction of ethyl pyruvate with R3N+âH is much stronger when ethyl pyruvate adopts an s-cis conformation due to strong ionâdipole interaction. This type of interaction between ethyl pyruvate and protonated cinchonidine is considered to be crucial for the enantio-differentiation in the heterogeneous enantioselective hydrogenation of ethyl pyruvate over cinchonidine modified platinum in acidic media. | ||||||||
|
||||||||
IR and NMR experiments revealed that the enantioselective hydrogenation of ethyl pyruvate in nonacidic solvents is complicated by the simultaneously occurring self-condensation (aldol reaction) of the reactant. Both enantioselective reactions are catalyzed by the chiral base cinchona alkaloid, but the hydrogenation is faster by several orders of magnitude than the aldol reaction. Catalytic experiments proved that the aldol products are not spectator species. The enol form of the major aldol product protonates the quinuclidine N of cinchonidine and enhances the enantiomeric excess of the hydrogenation reaction. The significance of this observation with respect to kinetic and mechanistic studies is discussed. | ||||||||
|
||||||||
Cinchona alkaloids play a major role as chiral auxiliaries in asymmetric catalysis. Acetic acid is known to be an excellent solvent in the enantioselective hydrogenation over chirally modified platinum metals. The crucial interaction between the chiral auxiliary and the solvent has been investigated using the cinchonidineâacetic acid pair. Solutions containing cinchonidine and acetic acid were studied by means of NMR and IR spectroscopy as well as by ab initio HartreeâFock calculations. In the presence of the acid cinchonidine is protonated at the quinuclidine N and adopts an open conformation where the quinuclidine N points away from the quinoline moiety. In the most stable 1â¶1 and 2â¶1 acetic acidâcinchonidine complexes both the NâH+ and OâH groups of cinchonidine are involved in hydrogen bonding. The most stable 1â¶1 complex is found to be cyclic. The relative arrangement of the NâH+ and OâH groups of protonated cinchonidine is ideally suited to bind an acetate anion, and the interaction hardly affects the cinchonidine conformation. Several 2â¶1 acidâbase complexes coexist in solution. The IR spectra give evidence for the existence of a 2â¶1 cyclic complex. Calculated structures, relative energies and vibrational frequencies are in good agreement with the experiment. The findings rationalise the importance of the OâH group of cinchonidine for the enantiodifferentiation in the enantioselective hydrogenation of α,β-unsaturated carboxylic acids over cinchonidine-modified Pd. | ||||||||